Avant d’être commercialisé, un médicament est d’abord évalué sur un échantillon de personnes dans le cadre d’essais cliniques pour mettre en évidence les effets indésirables et les risques encourus par les personnes. Tout ne pouvant être anticipé, en particulier ce qui concerne la prise au long cours, l’évaluation du médicament commercialisé se poursuit donc en une surveillance qu’on appelle pharmacovigilance après son obtention d’ATU ou d’AMM. Décodage de ce dispositif. Premier épisode.
Pharmacovigilance : définition
L’Organisation Mondiale de la Santé (OMS) définit la pharmacovigilance comme « une science ou un ensemble d’activités relatives à la détection, l’évaluation, à la compréhension et à la prévention des risques d’effets indésirables (ou de tout autre problème) liés aux médicaments mis sur le marché à titre onéreux ou gratuit (intolérance aux médicaments, mésusage ou usage abusif, erreur thérapeutique, pharmacodépendance, antibiorésistance, effets sur la femme enceinte ou sur l’enfant, échec thérapeutique) ». L’Agence française de sécurité sanitaire des produits de santé (Afssaps) précise que cette surveillance s’applique à un « risque soit potentiel ou avéré, des médicaments lorsqu’ils sont consommés largement dans le cadre de leur commercialisation ». La pharmacovigilance est indispensable pour assurer la sécurité des personnes. Les scandales du sang contaminé, ou plus récemment du médiator, montrent que des défaillances et des négligences au niveau de la pharmacovigilance peuvent avoir des conséquences dramatiques, voire criminelles.Surveiller un médicament, ça donne quoi dans la vraie vie ?
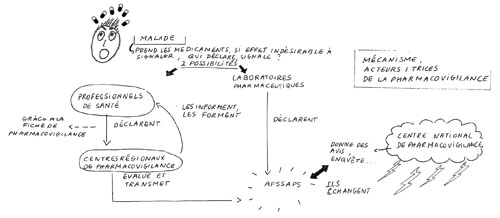
Les acteurs/actrices du système institutionnelLe mécanisme de pharmacovigilance s’articule entre les professionnelLEs de santé, les centres régionaux de pharmacovigilance et l’Afssaps.
- Les professionnelLEs de santé (médecins, pharmacienNEs, sages-femmes et chirurgienNEs-dentistes) doivent signaler tout « effet indésirable grave ou inattendu, susceptible d’être dû à un médicament », « qu’il l’ait ou non prescrit » à un centre régional de pharmacovigilance.
- Les centres régionaux de pharmacovigilance recueillent les signalements et les transmettent à l’Afssaps. Ils doivent informer et participer à la formation des professionnelLEs de santé en matière de pharmacovigilance. On en compte 31 (liste disponible sur le site de l’Afssaps).
- L’Afssaps enregistre et évalue ce qui lui est signalé. Une nouvelle notification[[nouvelle notification : par exemple, un nouvel effet indésirable listé dans la notice d’un médicament et qui donne lieu à une révision de l’AMM]] peut alors être envisagée ou un médicament supprimé du marché.
Comme les professionnelLEs de santé (ils ont parfois la double casquette) les industriels doivent signaler tout effet indésirable dû à un médicament dont ils ont connaissance et ce, en avertissant l’Afssaps dans un délai maximum de 15 jours. Les laboratoires doivent également soumettre à l’Agence des rapports périodiques actualisés de pharmacovigilance. Les laboratoires désignent unE interlocuteur/trice pour l’Afssaps, qui doit être médecin ou pharmacienNE. Son rôle est d’alerter l’Afssaps pour tout problème lié à la sécurité du médicament. Industrie pharmaceutique : une source d’information nécessairement biaisée ?
On voit bien les enjeux que peuvent soulever tout signalement d’effet indésirable supplémentaire d’un produit par un laboratoire. Notifier un nouvel effet indésirable, c’est faire éventuellement baisser le rapport bénéfice/risque pour les malades, et par conséquent, que ce produit soit moins prescrit et/ou moins remboursé par la sécurité sociale. Un laboratoire tient compte de la sécurité des personnes, avec en arrière plan, très proche, des objectifs de compétitivité et de profit, aussi bien avant (essais cliniques), qu’après l’AMM. Le Centre National de Pharmacovigilance (CNP)
Après évaluation des effets indésirables d’un médicament, les avis du CNP concernant les mesures à prendre sont transmis au ministère de la Santé et à l’Afssaps, il peut demander le retrait du marché d’un médicament. Il a également pour mission de proposer à l’Afssaps des études et enquêtes qu’il estime nécessaires.
Le CNP est composé de 6 membres de droit : la Direction Générale de la Santé, l’Afssaps, la Direction Générale de l’Offre de Soins, l’INSERM, la Commission nationale des stupéfiants et des psychotropes et la Commission nationale de pharmacovigilance vétérinaire. 33 membres sont désignéEs pour 3 ans par le ministère de la Santé, parmi lesquels : pharmacienNEs, médecins et unE représentantE de l’industrie pharmaceutique. Les malades et les associations La loi du 4 mars 2002, relative aux droits des malades et à la qualité des soins, a inscrit les malades en acteurs/actrices du système de Santé. En ce qui concerne la pharmacovigilance, cela est resté cependant au stade du souhait, puisque les malades eux/elles-mêmes ne peuvent signaler directement un effet indésirable. Cependant, comme l’indique l’Afssaps elle-même, les malades, en passant par unE médecin peuvent faire ce signalement (voir modalités ci-après). Les associations s’y emploient de leur côté avec les différentEs interlocuteurs/trices concernéEs (ministère de la Santé, Afssaps, laboratoires).
Signaler des effets indésirables
La déclaration se fait à partir de la fiche de pharmacovigilance[[Vous ne pouvez directement alerter l’Afssaps quant à la survenue d’effets indésirables que vous constatez, c’est au/à la professionnelLE de santé de documenter une fiche de pharmacovigilance, disponible sur le site de l’Afssaps et en pièce jointe de cet article).]]. En la remplissant et en la documentant, les professionnelLEs de santé peuvent alerter le Centre Régionale dont ils/elles dépendent, qui à son tour transmettra à l’Afssaps. Mais cette fiche est trop souvent méconnue des professionnelLEs de santé. Alternative possible pour unE professionnelLE de santé : prévenir le laboratoire, mais on est alors moins sûr du cheminement de l’information jusqu’à l’Afssaps.Que déclare-t-on ?
Selon l’Afssaps : – « Tout effet indésirable grave (létal, ou susceptible de mettre la vie en danger, ou entraînant une invalidité ou une incapacité importante ou durable, ou provoquant ou prolongeant une hospitalisation ou se manifestant par une anomalie ou une malformation congénitale). – Tout effet inattendu (dont la nature, la sévérité ou l’évolution ne correspondent pas aux informations contenues dans le RCP[[RCP : Résumé des Caractèristiques d’un Produit. Ce document doit obligatoirement accompagner tout médicament quelle que soit sa présentation galénique. Il en précise les caractéristiques, la posologie selon l’âge et la pathologie.]]). Mais aussi tout effet que vous jugez pertinent de déclarer en dehors de ces définitions. » Cette dernière phrase de l’Afssaps est une bonne synthèse : on déclare dès que ça paraît pertinent et l’on espère que la fiche sera enregistrée, évaluée et discutée, qu’elle ne se perdra pas dans une pile de dossiers à traiter. Lenteur administrative et effets indésirables constatés s’articulent mal.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}